心脏肥厚是全球发病率和死亡率的主要原因。肥厚过程部分由 Rho 家族的小 G 蛋白介导。我们假设他汀类药物,3-羟基-3-甲基戊二酰辅酶 A 还原酶的抑制剂,通过阻断 Rho 异戊烯基化来抑制心脏肥厚。我们用或不用辛伐他汀(Sim)处理新生大鼠心脏肌细胞,并发现 Sim 降低了 AngII 诱导的蛋白质含量、[3H]亮氨酸摄取和心房利钠肽(ANF)启动子活性。这些作用与细胞大小、膜 Rho 活性、超氧阴离子(O2·-)产生和细胞内氧化的降低有关,并且可以用 L-异戊二烯酸或香叶基香叶基焦磷酸酯逆转,但不能用法尼基焦磷酸酯或胆固醇逆转。使用 Rho 抑制剂 C3 外毒素和细胞渗透性超氧化物歧化酶的治疗也降低了 AngII 诱导的 O2·-产生和肌细胞肥厚。 过表达显性负性 Rho 突变体 N17Rac1 完全抑制了 AngII 诱导的细胞内氧化和ANF启动子活性,而 N19RhoA 部分抑制了它,N17Cdc42 则没有作用。实际上,辛伐他汀抑制了 AngII 灌注治疗或经主动脉缩窄处理的鼠的心脏肥大,并降低了心肌 Rac1 活性和 O2·̄的产生。这些发现表明,他汀类药物通过抑制 Rac1 的抗氧化机制预防心脏肥大的发展。
一、引言
心肌肥大是对血压升高或后负荷增加的初始生理适应性反应。然而,尽管通过药物使全身血压恢复正常,心肌肥大仍常会失代偿为充血性心力衰竭。事实上,心肌肥大是心血管疾病的独立危险因素,会使心血管死亡率增加两倍以上。肥大过程的特征包括诱导即刻早期基因(如c-fos、c-jun和erg-1)的表达;重新表达胚胎基因(如心房钠尿因子(ANF)、β-肌球蛋白重链和骨骼α-肌动蛋白);以及增加收缩蛋白(如肌球蛋白轻链-2(MLC-2v)和心肌α-肌动蛋白)的合成。
血管紧张素II(AngII)类型I受体的刺激或压力过载暴露可诱导心肌肥大反应,部分由异源三聚体G蛋白(例如,Gq)和小G蛋白(例如,Ras和Rho)的激活介导。特别是,Rho蛋白RhoA、Rac1和Cdc42被认为在调节细胞形态和收缩元素的过程中在肥大过程中发挥关键作用。例如,先前的研究表明,RhoA对于心肌细胞中的Gαq和α1-受体信号是必需的,并且RhoA的显性负突变体可以减轻对苯肾上腺素刺激的心肌细胞肥大。令人惊讶的是,在老鼠中,RhoA在心脏中的特异性过表达并不会导致心肌肥大,但会产生窦房结和房室结功能障碍以及心室衰竭的发展。这些结果表明,其他途径可能参与了肥大反应。事实上,Rac1而不是RhoA对于导致心肌细胞肥大的信号转导途径是必需的。
当前治疗心肌肥大的方法仅限于血管扩张剂或后负荷降低剂,很少有针对心肌过程的疗法。3-羟基-3-甲基戊二酰辅酶A(HMG CoA)还原酶抑制剂,或称他汀类药物,是广泛使用的降胆固醇药物,可降低心肌梗死和缺血性卒中的发生率。除了抑制胆固醇合成外,他汀类药物还抑制了合成重要的异戊二烯类中间体,这些中间体对于包括Rho蛋白在内的多种蛋白质的亚细胞定位和功能至关重要。因此,本研究旨在确定他汀类药物是否可以通过抑制Rho蛋白来减轻心肌肥大,如果是的话,确定涉及的潜在机制。
二、方法
2.1 心肌细胞培养
我们从1日龄的Sprague-Dawley大鼠中获取心室,并通过胰蛋白酶-EDTA和2型胶原酶消化分离心肌细胞。细胞在无血清的胰岛素-转铁蛋白(IT)培养基中培养24-36小时。使用这种方法,我们获得了超过95%的心肌细胞原代培养,通过显微镜观察自发细胞收缩和抗心肌肌球蛋白重链的免疫荧光染色来评估。除非另有说明,所有试剂均来自Sigma Chemical Co.,并使用以下最终浓度:AngII,1 μM;预激活的辛伐他汀,0.1-10 μM;L-甲羟戊酸(L-mev),200 μM;法尼基焦磷酸(FPP),10 μM;香叶基焦磷酸(GGPP),10 μM;低密度脂蛋白胆固醇,1 mg/ml;法尼基转移酶抑制剂,20 nM;香叶基转移酶抑制剂,30 μM;肉毒杆菌C3转移酶,50 μg/ml;聚乙二醇-超氧化物歧化酶(PEG-SOD),50 U/ml;聚乙二醇-过氧化氢酶(PEG-CAT),500 U/ml;和聚乙二醇(PEG),0.32 mg/ml。PEG、PEG-SOD和PEG-CAT溶解在IT培养基中,并在AngII刺激前与心肌细胞孵育1小时。心肌细胞刺激24小时。通过细胞数量、收缩频率(即固有心率)、细胞形态和台盼蓝排斥来确定细胞活性。
2.2 心肌肥大的测量
我们按照描述的方法测量[3H]亮氨酸摄取。心肌细胞在无血清的IT培养基中孵育36小时,然后在存在稀释剂或[3H]亮氨酸(1 μCi/ml)的条件下处理24小时。在室温下孵育45分钟后,用5%三氯乙酸沉淀细胞蛋白,用0.4 N NaOH重新悬浮,并在闪烁计数器中计数放射性。
2.3 北方印迹
将等量的总RNA(15 μg)通过1%甲醛-琼脂糖凝胶电泳分离,按照先前描述的方法进行杂交和洗涤。ANF和MLC-2v cDNA用随机六聚体引物和[α-32P]CTP(NEN Life Science Products Inc.,Boston, Massachusetts, USA)以及Klenow(Amersham Pharmacia Biotech Inc.,Piscataway, New Jersey, USA)进行标记。通过28S核糖体RNA的溴化乙锭染色确定装载条件。通过激光密度计分析印迹。
2.4 瞬时转染
接近汇合的鼠心肌细胞使用钙磷酸共沉淀法转染指定的cDNA。使用以下量的cDNA:突变的c-myc标记的RhoA、Rac1和Cdc42(各10 μg);ANF-Luc报告质粒(15 μg);和β-半乳糖苷酶质粒(4 μg)。转染后大约48小时,心肌细胞按照指示再处理24小时。将荧光素酶活性标准化为每个样本的β-半乳糖苷酶活性,以校正转染效率的差异。结果表示为相对于对照的相对比率(倍数诱导)。初步研究表明,转染效率约为2-4%。
2.5 免疫荧光
在转染和在指定条件下刺激后,鼠心肌细胞用10 μg/ml的二氯荧光素二乙酸酯处理1分钟,然后用1%甲醛固定并用0.1% Triton X-100通透化。对于c-myc标签的染色,细胞在37°C下用小鼠抗人c-myc抗体(1:100在1% BSA中)孵育20分钟。细胞用1% BSA洗涤后,使用R-藻红蛋白结合的山羊抗小鼠IgG(红色荧光)作为二抗(1:100在1% BSA中)。对于α-肌动蛋白和结蛋白的染色,细胞用小鼠单克隆抗体针对α-肌动蛋白(1:200在1% BSA中)和兔多克隆抗体针对结蛋白(1:100在1% BSA中.)孵育。细胞用1% BSA在PBS中洗涤后,用这些二抗处理:FITC结合的山羊抗小鼠IgG(H+L)(绿色荧光)和TRITC结合的抗兔IgG(H+L)(红色荧光)(1:200在1% BSA中)。使用配备氪氩和钛宝石激光器的MRC-1024/2P多光子显微镜观察免疫荧光。从五个随机视野拍摄照片。
2.6 Rho GTP结合活性
我们通过免疫沉淀[35S]GTP-γS标记的Rho蛋白来测量培养的心肌细胞或鼠心脏中膜相关的Rho GTP结合活性,使用部分纯化的心肌细胞膜和特异性RhoA和Rac1抗体。免疫沉淀后的上清液经免疫印迹分析,结果显示RhoA和Rac1被完全沉淀。在过量未标记的GTP-γS(100 μM;NEN Life Science Products Inc.)存在下测定非特异性活性。
2.7 铁细胞色素c还原试验
通过测量铁细胞色素c的超氧化物歧化酶(SOD)可抑制的还原来测定培养上清液中的超氧阴离子(O₂⁻)。在无酚红和血清缺乏的IT培养基中刺激24小时后,将铁细胞色素c加入上清液中,使其终浓度达到70 μM,同时存在或不存在SOD(100 U/ml)。使用分光光度计在550 nm处监测铁细胞色素c的还原,持续10分钟。按照描述的方法计算O₂⁻的生成速率,结果以每百万细胞每24小时生成的纳米摩尔数表示。
2.8 心脏组织中O₂⁻的生成测定
将大鼠或小鼠心脏(100-200 mg)的完整、未匀浆的组织块悬于Krebs碳酸氢盐缓冲液中,其中包含以下试剂,单位为mmol/l:NaCl,118;KCl,4.7;CaCl₂,1.5;MgSO₄,1.1;KH₂PO₄,1.2;葡萄糖,5.6;NaHCO₃,25。将该组织浴调至pH 7.4,并用21% O₂和5% CO₂通气4小时。使用上述方法测定上清液中铁细胞色素c的SOD可抑制还原。结果以每克心脏组织产生的O₂⁻的纳米摩尔数表示。
2.9 醋酸酶活性测定
通过醋酸酶活性的变化间接测定细胞内O₂⁻的生成。将培养细胞溶于含有PBS、Triton X-100(0.2%)、DTPA(100 mM)和柠檬酸(5 mM)的裂解缓冲液中。将心脏组织匀浆并重悬于含有Tris-HCl(50 mM,pH 7.6)、半胱氨酸(1 mM)、柠檬酸(1 mM)和MnCl₂(0.5 mM)的缓冲液中。将15 μg的细胞提取物加入含有Tris-HCl(50 mM,pH 7.4)、异柠檬酸(20 mM)和MnCl₂(0.5 mM)的反应缓冲液(0.2 ml)中,在25°C下,通过分光光度计在240 nm处测量2分钟后顺式阿康酸的生成。使用3.6 mM⁻¹×cm⁻¹的消光系数计算醋酸酶活性,并以每分钟每毫克蛋白转化的顺式阿康酸的纳米摩尔数表示。
2.10 心肌肥大的动物模型
我们使用了8周大的雄性Sprague-Dawley大鼠(200-250 g),来自Taconic Farms。大鼠通过渗透微型泵接受生理盐水(25 μl/h)或AngII(200 ng/kg/min),有无1.0 ml的预激活辛伐他汀(0.2, 2, 或20 mg/kg/day,皮下注射)或相应体积的PBS,持续14天。使用尾袖法(IITC Inc.,Woodland Hills, California, USA)非侵入性测量收缩压。在治疗前后,分别测定左心室(LV)质量、体重(BW)和血清总胆固醇水平。
对于压力过载模型,我们使用了8周大的雄性C57BL/6小鼠(20-25 g),来自Charles River Laboratories。通过用7-0缝线在主动脉周围缠绕两次并插入27号针头来创建主动脉缩窄(TAC)。然后轻轻抽出针头,产生60-80%的缩窄,主动脉外径约为0.3 mm。小鼠在手术后立即接受载体或预激活辛伐他汀(2 mg/kg/day,皮下注射)。TAC 4周后,小鼠用腹腔注射戊巴比妥(50 mg/kg)麻醉,通过15-MHz脉冲波多普勒超声心动图分析心脏尺寸和功能。通过左颈动脉侵入性测量系统动脉血压。
三、结果
3.1 细胞培养
通过相位接触显微镜观察形态特征和用单克隆抗心肌肌球蛋白重链抗体进行免疫荧光染色,证实了相对纯度(>95%)的新生大鼠心室肌细胞(数据未显示)。在任何处理条件下,AngII、Sim、L-mev、LDL、GGPP、FPP、GGTI-286、FTI-276、C3 TF、PEG-SOD或PEG-CAT对细胞活性均无明显不良影响。
3.2 他汀类药物抑制AngII诱导的心肌细胞肥大
心肌细胞肥大以蛋白含量增加(例如,[3H]亮氨酸摄取)和胎儿基因表达诱导(例如,ANF启动子活性)为特征。AngII处理诱导新生大鼠心肌细胞肥大,通过[3H]亮氨酸摄取、总蛋白含量和ANF启动子活性的变化来衡量(分别增加了50%±6%、20%±3%和180±20倍;所有条件P<0.01)(图1a)。与辛伐他汀(5 μM)联合处理抑制了AngII诱导的[3H]亮氨酸掺入、总蛋白含量和ANF启动子活性的增加,分别抑制了71%±4%、85%±8%和94%±5%。这些辛伐他汀的抑制效应完全被L-mev或GGPP联合处理逆转,但FPP或LDL联合处理则无此效应(图1)。然而,单独使用辛伐他汀处理并未影响基础的[3H]亮氨酸掺入、总蛋白含量或ANF启动子活性。其他他汀类药物(例如,洛伐他汀、阿托伐他汀)也观察到类似的效果,表明这些药物对心肌HMG CoA还原酶具有类别效应。
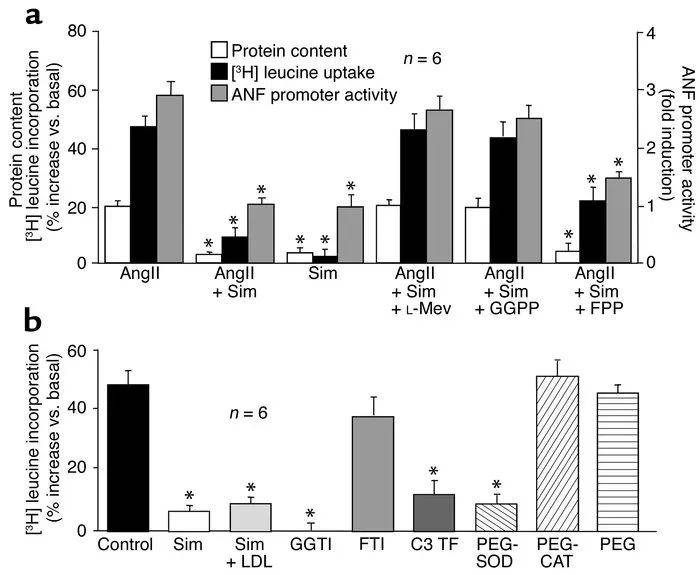
图1. 他汀类药物对心肌细胞肥大的抑制作用。(a)AngII或辛伐他汀(5 μM)与L-mev、GGPP或FPP联合处理对总蛋白含量、[3H]亮氨酸掺入和ANF启动子活性的影响。数值以均值±标准误表示。与单独使用AngII相比,P<0.01。(b)辛伐他汀(5 μM)与LDL、GGTI、FTI、C3 TF、PEG、PEG-SOD和PEG-CAT联合处理对AngII诱导的[3H]亮氨酸掺入的影响(对照)。数值以均值±标准误表示。与对照相比,P<0.01。
使用选择性香叶基化和法尼基化抑制剂在其最佳抑制浓度下,我们发现GGTI-286(30 μM)完全阻断了AngII诱导的[3H]亮氨酸掺入,而FTI-276(20 nM)则无此效应(图1b)。为了确定辛伐他汀对心肌细胞肥大的影响是否通过抑制Rho蛋白介导,我们直接用C3 TF(50 μg/ml)抑制Rho蛋白。C3 TF处理抑制了AngII诱导的[3H]亮氨酸掺入增加59%±5%。相比之下,通过大肠杆菌细胞毒素坏死因子-1直接激活Rho部分逆转了辛伐他汀的抑制效应(数据未显示)。由于Rho蛋白Rac1参与活性氧(ROS)的生成,并介导导致心肌肥大的信号通路,我们研究了超氧阴离子(O₂⁻)或过氧化氢(H₂O₂)是否在AngII诱导的心肌肥大中起作用。与细胞可渗透的PEG-SOD联合处理,而非PEG-CAT或PEG单独处理,抑制了AngII诱导的[3H]亮氨酸掺入(图1b)。这些结果表明,肥大过程更多地依赖于O₂⁻而非H₂O₂。

东莞市富临塑胶原料有限公司是IITC神经科学设备中国代理商,为中国客户提供神经科学产品:大小鼠血压测量系统、足底测痛仪、疲劳转棒仪、大小鼠跑步机。
为了确定辛伐他汀是否影响心肌细胞大小和肌节组织,我们对心肌细胞进行了针对两种肌节相关蛋白——结蛋白和α-肌动蛋白的免疫染色。AngII处理诱导细胞大小和肌节组织显著增加(图2a);与辛伐他汀联合处理阻断了这些AngII诱导的细胞变化。肥大过程还与胎儿和结构性心脏基因如ANF和MLC-2v的诱导有关。实际上,经AngII刺激后,ANF和MLC-2v mRNA的稳态表达均增加了约两倍(n=3,P<0.01)(图2,b和c)。与辛伐他汀联合处理抑制了心肌细胞中AngII诱导的ANF以及在较小程度上的MLC-2v mRNA表达。高基础表达的ANF和MLC-2v归因于使用新生心肌细胞而非成人心肌细胞。尽管如此,这些结果与辛伐他汀对心肌细胞肥大的抑制作用相关。
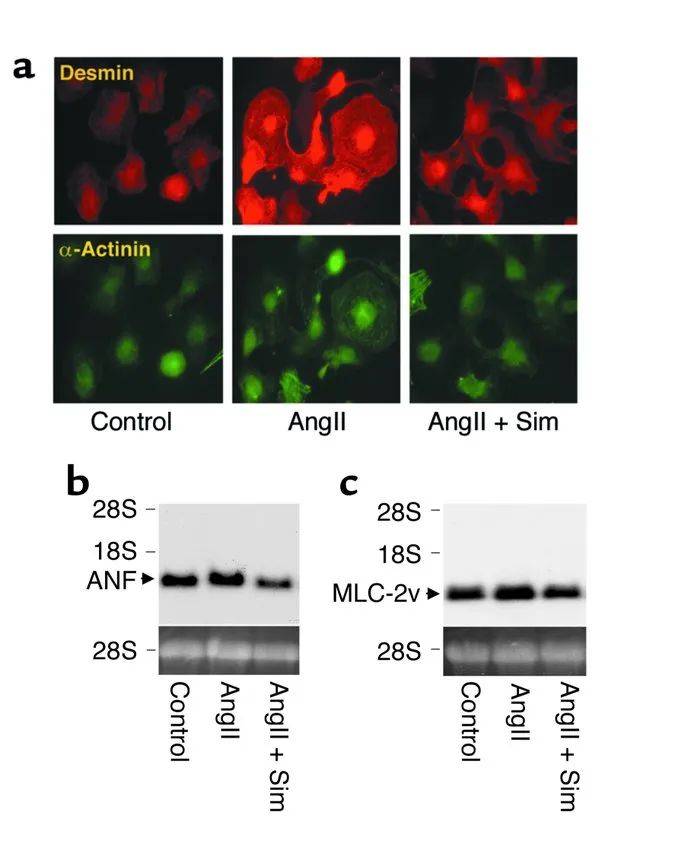
图2. 他汀类药物对心肌细胞肌节组织、细胞大小和胎儿基因表达的抑制作用。(a)AngII与辛伐他汀(5 μM)联合处理对心肌细胞大小和肌节组织的影响。采用特异性抗体对结蛋白(上图,红色)和α-肌动蛋白(下图,绿色)进行双重免疫荧光显微镜观察。实验重复三次,结果相似。AngII与辛伐他汀(5 μM)联合处理对经24小时处理后的(b)ANF和(c)MLC-2v mRNA稳态表达的影响。以溴化乙锭染色的28S核糖体RNA作为标准化标准。所示结果为三次独立实验的代表。
3.3 他汀类药物对Rac1和RhoA的抑制作用
AngII处理使膜相关RhoA和Rac1的GTP结合活性分别增加了147%和87%(两者P<0.05)(图3a)。与辛伐他汀联合处理完全阻断了AngII诱导的膜相关RhoA和Rac1 GTP结合活性的增加。这些辛伐他汀的抑制效应被GGPP联合处理逆转,而非FPP,表明辛伐他汀抑制了RhoA和Rac1的膜定位和活性。
为了评估哪种Rho蛋白介导AngII诱导的心肌肥大,我们将大鼠心肌细胞共转染了Rac1、RhoA或Cdc42的显性负突变体以及ANF启动子-荧光素酶报告构建体。转染显性负Rac1(N17Rac1),以及在较小程度上转染RhoA(N19RhoA),抑制了AngII诱导的ANF启动子活性(图3b)。转染Cdc42(N17Cdc42)则无抑制效应。与辛伐他汀联合处理进一步降低了转染N19RhoA和N17Cdc42的细胞中的ANF启动子活性,但对转染N17Rac1的细胞则无此效应。实际上,N17Rac1将ANF启动子活性降至基础水平以下(P<0.05)。尽管GGPP联合处理逆转了辛伐他汀的抑制效应,但GGPP无法逆转N17Rac1对ANF启动子活性的抑制效应。这些结果表明,Rac1是介导AngII诱导的心肌肥大的主要Rho蛋白。
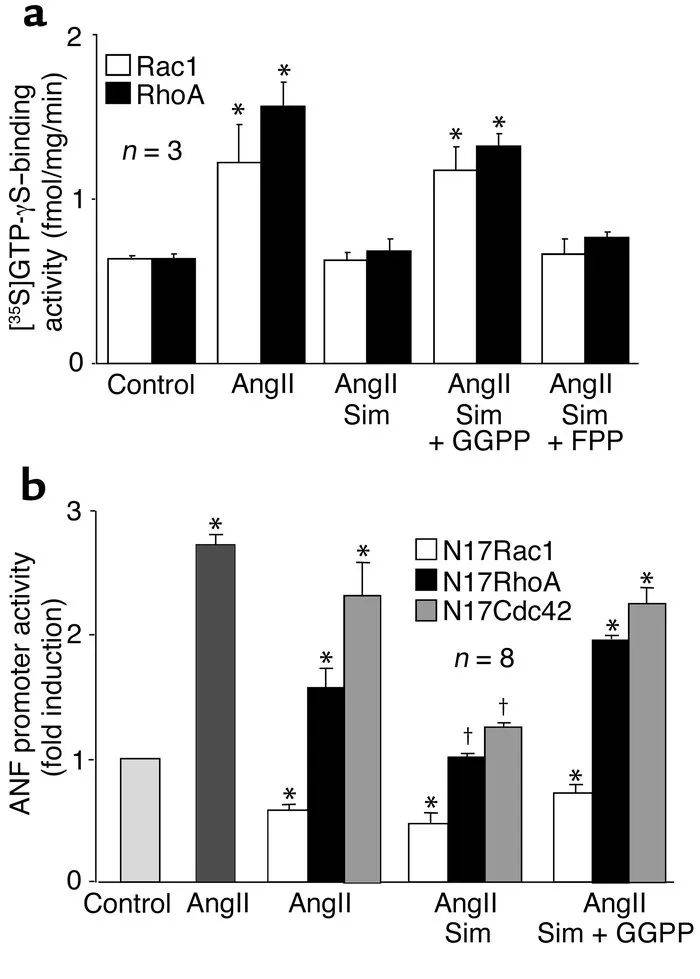
图3. 他汀类药物对RhoA和Rac1的抑制作用。(a)AngII与辛伐他汀、GGPP或FPP联合处理对大鼠心肌细胞膜相关Rac1和RhoA GTP结合活性的影响。数值以均值±标准误表示。与未刺激细胞(对照)相比,P<0.05。(b)转染显性负Rho突变体(N17Rac1、N19RhoA或N17Cdc42)与辛伐他汀(5 μM)或GGPP联合处理对AngII诱导的ANF启动子活性的影响。数值以均值±标准误表示。与仅转染载体(对照)相比,P<0.01。†P<0.05,与AngII和显性负Rho单独处理相比。Tx:DN-Rho=N17Rac1、N19RhoA或N17Cdc42。
3.4 他汀类药物抑制Rac1诱导的O₂⁻生成
Rac在中性粒细胞、成纤维细胞和血管平滑肌细胞中的一个重要功能是促进NADPH氧化酶的组装,这是这些细胞中O₂⁻生成的主要来源。为了确定辛伐他汀对心肌肥大的抑制效应是否涉及抑制O₂⁻生成,我们采用铁细胞色素c还原试验监测心肌细胞释放到上清液中的O₂⁻。AngII使O₂⁻生成量增加一倍(图4a)。这种增加被与辛伐他汀、C3 TF和PEG-SOD(50 U/ml)联合处理阻断,而PEG-CAT(500 U/ml)联合处理则无此效应。单独使用PEG或辛伐他汀处理对O₂⁻生成影响甚微或无影响。与GGPP联合处理完全逆转了辛伐他汀对O₂⁻生成的抑制效应,但对C3 TF或PEG-SOD的效应则无逆转作用(数据未显示)。
为了确认AngII和辛伐他汀对O₂⁻生成的影响是否对应于细胞氧化应激的变化,我们通过DCF荧光评估了大鼠心肌细胞中的总细胞内氧化。由于H₂O₂是O₂⁻的下游歧化产物,DCF荧光水平与过氧化氢衍生的活性中间体水平增加相关,也可作为O₂⁻生成的有用标记。实际上,AngII诱导的DCF荧光被PEG-CAT的加入完全阻断,PEG-CAT将H₂O₂转化为H₂O,而PEG-SOD则无此效应,PEG-SOD增强了而非抑制了O₂⁻向H₂O₂的转化(数据未显示)。AngII刺激使细胞内氧化应激增加了2.7倍,通过DCF荧光测量(n=3,P<0.05)(图4b)。这种增加被与辛伐他汀联合处理完全阻断。辛伐他汀对细胞内氧化的抑制效应被L-mev和GGPP联合处理逆转,但FPP联合处理则无此效应,表明Rho蛋白介导了对AngII响应的O₂⁻衍生的细胞内氧化增加。
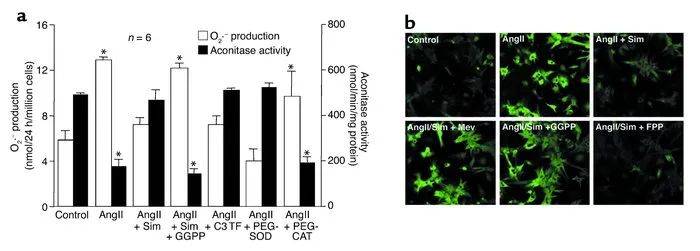
图4. 他汀类药物对超氧阴离子生成和细胞内氧化的抑制作用。(a)辛伐他汀(5 μM)、GGPP、C3 TF、PEG-SOD或PEG-CAT对大鼠心肌细胞中AngII诱导的O₂⁻生成的影响,通过铁细胞色素c还原和醋酸酶试验进行测量。数值以均值±标准误表示。与未刺激细胞(对照)相比,P<0.01。(b)辛伐他汀(5 μM)与L-mev、GGPP或FPP联合处理对AngII诱导的细胞内氧化(DCF荧光)的影响。所示结果选自五个随机视野,为三次独立实验的代表。
3.5 Rac1是AngII诱导的细胞内氧化所必需的
为了确定哪种Rho蛋白介导AngII诱导的细胞内氧化增加,我们将大鼠心肌细胞转染了c-myc标记的显性负(N17或N19)或组成性活性(L61或L63)Rho突变体,并对c-myc和细胞内氧化进行双重荧光显微镜观察。AngII处理增加了非转染细胞或转染了显性负RhoA或Cdc42的细胞中的细胞内氧化,通过DCF荧光确定(图5a)。然而,转染了显性负Rac1的细胞则完全抑制了AngII诱导的细胞内氧化。相反,只有转染了组成性活性Rac1的细胞显示出细胞内氧化增加;非转染细胞或转染了组成性活性RhoA或Cdc42的细胞则无此增加(图5b)。这些结果表明,Rac1介导了AngII诱导的细胞内氧化,并提示辛伐他汀通过抑制Rac1抑制了细胞内氧化。
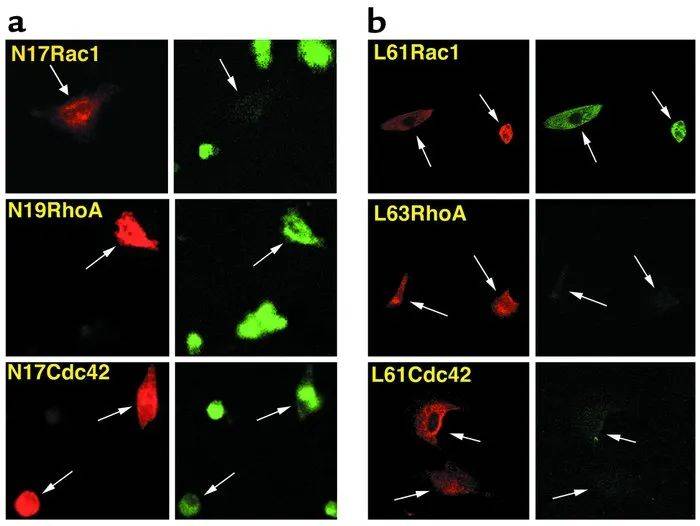
图5. 他汀类药物对Rac1诱导的细胞内氧化的抑制作用。转染了显性负Rho突变体(N17或N19)的大鼠心肌细胞经AngII刺激(a),或转染了组成性活性Rho突变体(L61或L63)(b)。采用双重荧光显微镜观察c-myc标记的Rho突变体的表达(左图,红色)和相应的DCF荧光水平(右图,绿色)。实验重复三次,结果相似。箭头指向相应图中的转染心肌细胞。
3.6 他汀类药物在体内抑制心肌肥大
为了确定我们的体外发现是否具有生理相关性,我们采用两种广泛使用的心肌肥大模型评估了他汀类药物的效应。AngII输注导致收缩压显著升高,这种升高未受辛伐他汀联合处理的影响(表1)。单独使用辛伐他汀处理对系统血压、体重或血清总胆固醇水平均无影响。AngII输注(200 ng/kg/天,持续14天)导致左心室/体重(LV/BW)比值和左心室质量显著增加。然而,在我们实验条件的短时间内,AngII并未导致胶原沉积或纤维化显著增加。AngII诱导的LV/BW比值和左心室质量的增加被辛伐他汀联合处理以浓度依赖性方式抑制。阿托伐他汀也观察到类似的结果,表明对心肌HMG CoA还原酶抑制具有类别效应。
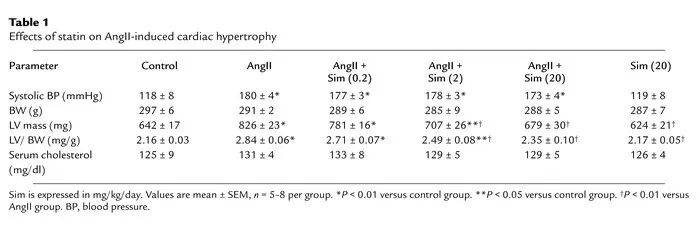
表1. 他汀类药物对AngII诱导的心肌肥大的影响
在经AngII输注处理的大鼠心脏中,膜相关RhoA和Rac1的GTP结合活性增加,这些活性均被辛伐他汀联合处理抑制(图6a)。实际上,AngII使完整、未匀浆的大鼠心脏组织中O₂⁻的生成增加了40%(从42±4增加到59±5 nmol/mg,n=6,P<0.05),这种效应被辛伐他汀联合处理完全阻断(34±8 nmol/mg,n=6,与对照相比P>0.05)(图6b)。这些结果与大鼠心脏组织中醋酸酶活性在AngII输注后降低了41%(从440±27降低到260±41 nmol/min/mg,n=6,P<0.005)相关,这种降低被辛伐他汀联合处理完全恢复(460±31 nmol/min/mg,n=6,与对照相比P>0.05)。这些结果表明,抑制Rho蛋白促成了他汀类药物对AngII诱导的O₂⁻生成的抑制效应。
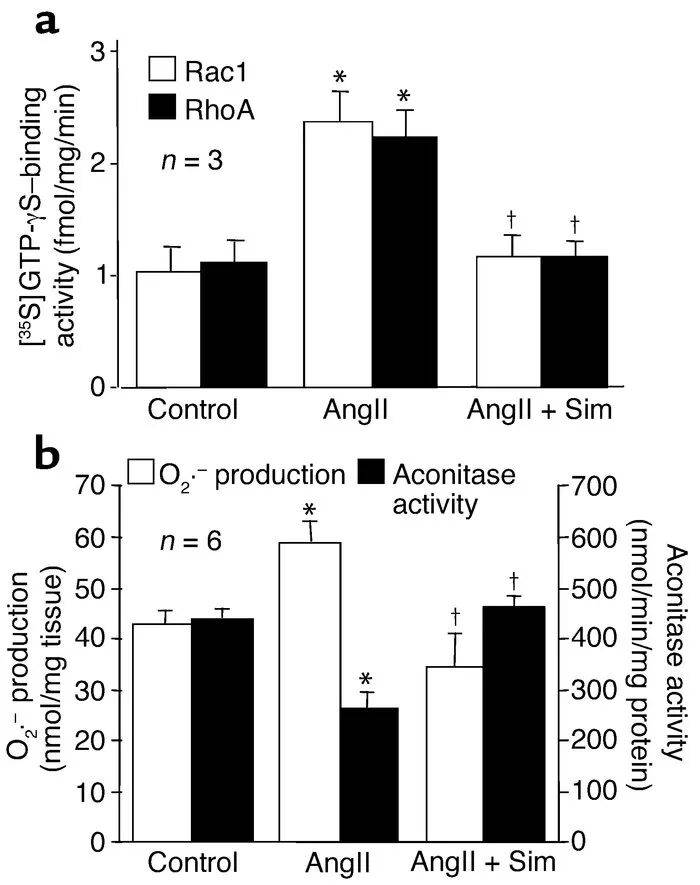
图6. 他汀类药物对大鼠心脏中Rac1和O₂⁻生成的抑制作用。AngII(200 ng/kg/min)输注与辛伐他汀(20 mg/kg/天,持续14天)联合处理对(a)膜相关Rac1和RhoA GTP结合活性以及(b)完整大鼠心脏中释放的O₂⁻和醋酸酶活性的影响。数值以均值±标准误表示。与载体处理动物(对照)相比,P<0.05。†P<0.05,与单独接受AngII处理的动物相比。
为了确定他汀类药物的抗肥大效应是否是一种可以推广到其他心肌肥大病因的核心机制,我们研究了辛伐他汀在压力过载心肌肥大模型中的效应。主动脉缩窄(TAC)并未影响收缩压或体重。然而,TAC导致心重/体重(HW/BW)比值显著增加,与假手术小鼠相比(图7a)。这些TAC的效应与小鼠心脏组织中醋酸酶活性降低了59%(从490±22降低到201±18 nmol/min/mg,n=3,P<0.01)相关。实际上,TAC导致了解剖学和功能学变化,这些变化与心肌肥大一致,如后壁厚度增加和左心室收缩末期直径增加,以及射血分数或缩短分数降低了25%(表2)。与辛伐他汀联合处理使HW/BW比值降低了约50%,完全恢复了小鼠心脏组织中的醋酸酶活性,并改善了心脏舒张功能,以及在较小程度上改善了收缩功能。此外,辛伐他汀处理显著降低了心肌细胞大小、左心室壁厚度和心脏整体尺寸(图7b)。
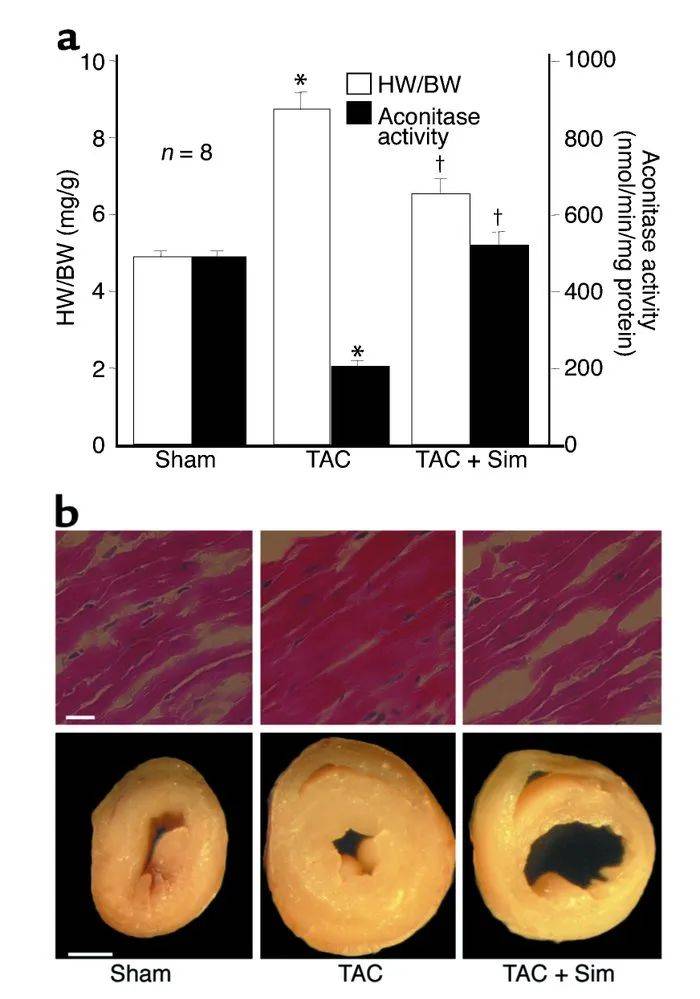
图7. 他汀类药物对压力过载诱导的O₂⁻生成和心肌肥大的抑制作用。(a)TAC与辛伐他汀(2 mg/kg/天,持续4周)联合处理对小鼠HW/BW比值和小鼠心脏中醋酸酶活性的影响。数值以均值±标准误表示。与接受载体处理的假手术动物(假手术)相比,P<0.01。†P<0.05,与未经辛伐他汀处理的TAC治疗相比。(b)通过苏木精-伊红染色对心脏组织(上排;条=10 μm)和在二尖瓣水平以下的死后心脏横截面宏观检查(下排;条=1 mm)评估心肌肥大。
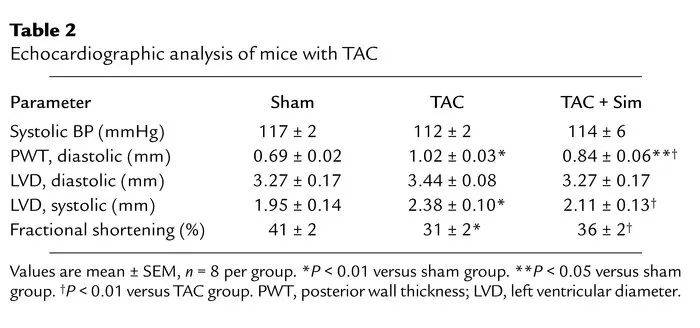
表2. 经TAC处理的小鼠的超声心动图分析
四、讨论
他汀类药物以胆固醇无关的方式预防心肌肥大的发展。其机制部分是由于抑制异戊二烯合成、Rho香叶基化和心肌细胞中Rac1诱导的O₂⁻生成。实际上,我们发现非特异性抗氧化剂N-乙酰半胱氨酸与他汀类药物一样有效地抑制了AngII诱导的心肌肥大(数据未显示)。然而,需要进一步研究以确定N-乙酰半胱氨酸抑制心肌肥大的确切机制,以及它是否涉及其抗氧化效应。尽管如此,他汀类药物通过抑制Rac1减少O₂⁻生成的能力表明,它们可能在减轻肥大过程方面具有超出降脂的临床益处。
有证据表明他汀类药物和抗氧化剂可能对心肌肥大具有抑制效应,尽管这些效应的机制尚不清楚。例如,最近的一项研究表明,他汀类药物可以在不影响Rac-GTP活性的情况下抑制兔子β-肌球蛋白重链-Q403突变模型中的心肌肥大和纤维化。这些结果表明,他汀类药物的抗肥大效应可能不仅仅由于抑制Rho蛋白,还可能涉及抑制其他下游信号通路。与其他研究相比,我们并未在AngII输注模型中观察到心脏纤维化增加,这可能是因为我们使用的AngII浓度较低且处理时间较短。实际上,使用更高浓度的AngII会观察到心肌肥大和纤维化。我们的发现与先前的一项研究一致,即他汀类药物通过抑制Rac诱导的O₂⁻生成减轻血管平滑肌细胞肥大。
他汀类药物的抗肥大效应也可能部分是由于降低了AngII类型1受体表达或心肌血管紧张素转换酶活性。然而,他汀类药物对其他激动剂(如内皮素-1和苯肾上腺素)以及压力过载诱导的心肌细胞肥大的抑制效应表明,主要作用机制包括AngII类型1受体下游的机制。他汀类药物治疗还增加了血管一氧化氮生成,这可能有助于降低血压并减轻肥大过程。然而,在我们的研究中,接受他汀类药物治疗的动物的系统性动脉收缩压并无显著变化。最后,一氧化氮也可能通过改善心脏性能和代谢的有利效应直接发挥他汀类药物的抗肥大效应。进一步在eNOS–/–小鼠中进行的他汀类药物研究应有助于澄清这些问题。
他汀类药物抑制了重要的异戊二烯中间体(如GGPP和FPP)的合成,这些中间体对于小GTP结合蛋白(如Rho)的翻译后修饰至关重要。Rho蛋白在介导心肌肥大的发展过程中发挥关键作用。例如,RhoA通过激活Rho激酶和肌球蛋白轻链磷酸化控制肌动蛋白应力纤维和粘着斑复合物的形成。Rac1和Cdc42调节称为片状伪足和丝状伪足的肌动蛋白细胞骨架过程,这些过程可能涉及与心肌肥大相关的形态学变化。此外,Rho蛋白还可以通过激活下游信号分子(如丝裂原活化蛋白激酶)来调节肥大过程。我们发现过表达显性负Rac1完全抑制了细胞内氧化和心肌细胞肥大,而RhoA则在较小程度上抑制,Cdc42则无抑制作用,这与先前的研究一致,即Rac1是心肌细胞肥大信号转导途径所必需的。
由于肥大反应是对后负荷增加的生理适应机制,因此尚不清楚以降低心脏性能为代价抑制肥大过程是否真的有益。例如,在左心室压力过载的实验模型中,抑制肥大反应会导致扩张型心肌病和心力衰竭的迅速发展。在心肌梗死后,如果抑制肥大过程或心肌重构,通常也会出现类似的有害效应。然而,尽管存在这些担忧,我们的结果表明,他汀类药物不仅减少了心肌肥大,还通过主要减少左心室舒张末期尺寸改善了心脏性能,即使在系统血压和后负荷持续升高的情况下也是如此。他汀类药物具有这些特性的未知确切机制,但可能涉及增强一氧化氮介导的心肌能量代谢改善。
总之,他汀类药物是预防心肌肥大发展的有效药物。具有临床意义的是,高血压而非高胆固醇是心肌肥大的主要危险因素。因此,这些结果表明了一种治疗心肌肥大的新药理学方法,适用于他汀类药物治疗可能不被推荐的患者群体。然而,在提出此类建议之前,需要来自大规模临床试验的支持数据。
东莞市富临塑胶原料有限公司是IITC中国代理商,采购IITC神经科学产品(大小鼠血压测量系统、足底测痛仪、疲劳转棒仪、大小鼠跑步机)请立即联系我们。

添加图片注释,不超过 140 字(可选)