
林奇样综合征的致病机制到底是啥?遗传易感基因检测分析其与林奇综合征和散发结直肠癌的分子特征
林奇样综合征(LLS)呈现出与林奇综合征(LS)极为相似的临床病理特征,但其癌症易感性机制仍不明确。本研究旨在通过全面的基因和表观遗传学方法探究LLS的因果机制。研究纳入了符合Amsterdam和Bethesda标准的32例LLS结直肠癌(CRC)患者、34例LS CRC患者以及29例散发性CRC患者,分析了94个与遗传性肿瘤相关基因的致病性变异情况。此外,研究还对队列的甲基化谱进行了表征,并通过样本组分析和随机表观遗传突变(SEMs)分析,与29名年龄匹配的健康对照进行了比较。多基因 panel 分析显示,非错配修复(MMR)基因中存在致病性变异,且有3个被分类为致病性/可能致病性的变异可能导致LLS。表观遗传学分析表明,表观变异发生在与LS或DNA修复相关的基因,其中大部分与范可尼贫血(Fanconi Anemia)通路相关,这可能解释了癌症易感性。本研究结果强调,需要通过扩展的基因和表观遗传学分析来理解LLS的因果机制。
研究背景
LLS是一种以CRC伴MSI和免疫组化(IHC)检测到MMR基因(MSH2、MLH1、MSH6、PMS2)之一表达缺失为特征的疾病。与LS不同,这两个特征并非由MMR基因的胚系突变引起,其因果机制仍不明确。LS和LLS的临床诊断需应用Amsterdam标准,并结合Bethesda标准。根据最新指南,所有CRC患者均需检测MSI和/或通过IHC检测MMR基因缺陷(dMMR),并可能检测BRAF V600E突变和MLH1启动子高甲基化。若检测显示MSI和/或dMMR(无BRAF突变和MLH1高甲基化),患者需接受遗传咨询,以评估家族史及个体临床和组织病理特征。这种筛查虽不足以确诊,但可识别高危患者。诊断路径的下一步是确定MMR基因是否存在潜在突变:当胚系检测确认致病性/可能致病性变异时,患者被诊断为LS;否则归类为LLS。约55%疑似LS的患者在基因检测后被归为LLS病例。LLS患者及其亲属的CRC风险升高(尽管低于LS患者),提示存在遗传因素。此外,LLS患者的CRC诊断年龄略高于LS患者(53.7岁 vs. 48.5岁),但仍低于散发性CRC患者。由于缺乏明确的因果机制,LLS患者被认为是一组具有LS和散发性CRC之间特征的异质性群体。MMR基因的双体细胞突变可解释部分LLS病例,但其常见于无癌症家族史的患者,因此应视为散发性CRC。LLS至少存在四种潜在病因:第一,MMR基因中仍存在意义未明变异(VUS),或第二,常规诊断方法未检测到的重排或调控区域变异等;第三,非MMR基因(如MUTYH、EXO1、POLE、POLD1、MCM8/9、WRN、BARD1、RCF1、RPA1、MLH3,以及近年发现的PPARG、CTC1、DCC、ALPK、PRKDC)的突变与LLS表型相关;第四,体质性表观遗传改变等额外机制可能导致MMR缺陷表型并遗传。尽管罕见,文献已报道MLH1和MSH2的体质性表观突变与LS相关,不排除其他基因的表观突变可能在LLS中起致病作用。
本研究旨在探索经遗传咨询筛选、已证实存在癌症易感性的LLS患者的潜在病因。研究者使用包含94个遗传性肿瘤高频突变基因的检测panel对LS、LLS 和散发性 CRC 患者的胚系DNA进行检测,并结合全面的表观遗传学特征分析,评估LLS与LS及散发性病例的潜在病因和基因/表观遗传相似性。
研究结果
患者特征:
本研究招募了LS、LLS、CRC患者及健康对照。LS队列包含32例根据Amsterdam和Bethesda指南诊断为LS的患者,且通过测序和拷贝数变异(CNV)分析确认MMR基因存在致病性变异。LLS队列包含34例符合Amsterdam和Bethesda标准但MMR基因无致病性变异的患者。散发性CRC队列包含29例诊断年龄>60岁且无LS相关癌症亲属的患者(表1)。此外,还招募了29名年龄匹配的健康对照(中位年龄:55岁,范围:35-67岁)用于表观遗传学分析。LS队列女性患者比例(73.5%)高于LLS队列(43.8%)和散发性CRC队列(34.5%)。各组在年龄和第二原发肿瘤发生率上存在显著差异:LS组肿瘤中位发病年龄为43岁,LLS组为56.5岁,散发性组为68岁;第二原发肿瘤发生率在LS组最高(68.7%),其次为LLS组(29.4%)和散发性组(6.9%)。LS患者中,31.3%出现第三原发肿瘤,6.2%出现第四原发肿瘤。LS组第二原发肿瘤好发于结肠(31.8%)和子宫内膜(22.7%),与文献和指南一致;LLS组则多见于结肠(40%)和乳腺(20%)。总体而言,LLS患者表现出介于LS和散发性CRC之间的特征。
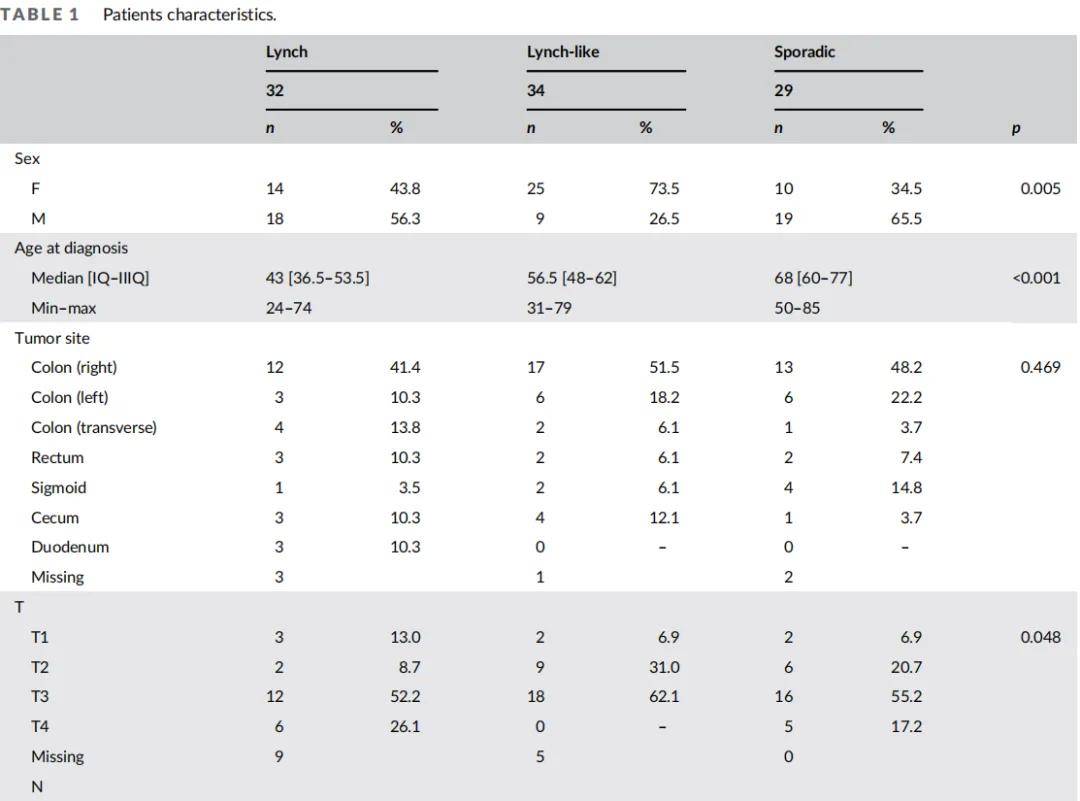
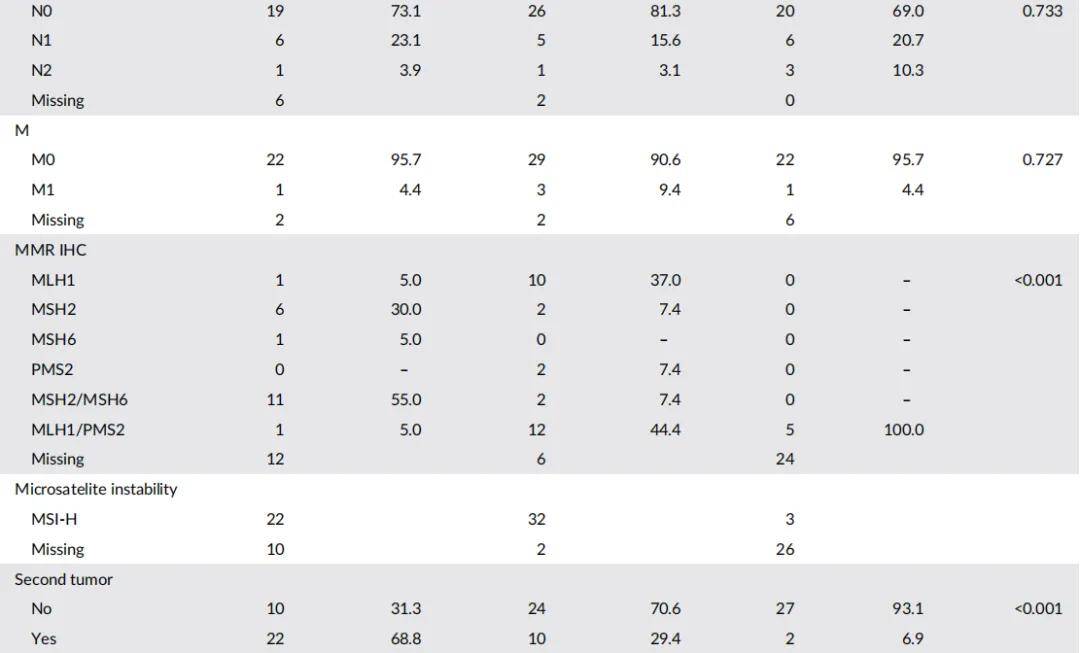
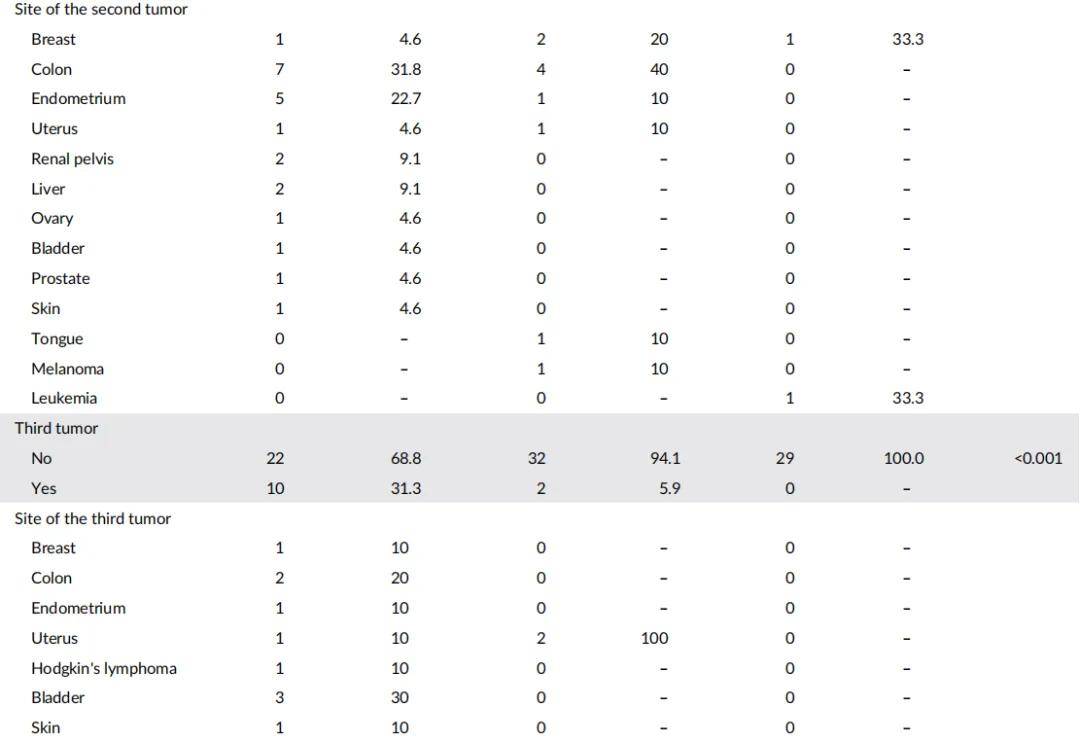
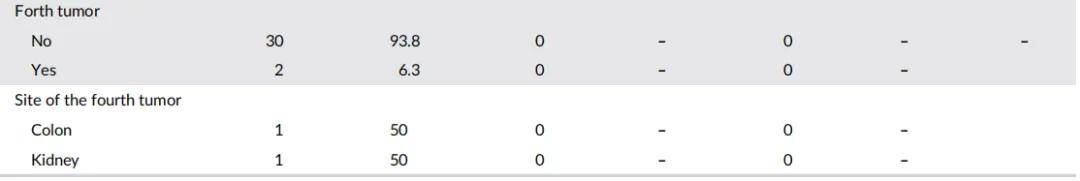
表1
LS、LLS和散发性CRC的基因组特征:
所有LS患者均表现为微卫星高度不稳定和/或MMRIHC异常染色,主要为MSH2或MSH2/MSH6表达缺失。94基因panel和CNV分析显示,LS队列中78%的MMR基因致病性变异基因为MSH2,其次为MLH1(16%)、EPCAM(6%)和MSH6(3%)。4例LS患者存在双重致病性变异(EPCAM/MSH2、ATM/MSH2、BUB1B/MSH2、PALB2/MSH2)。1例患者携带包含EPCAM和MSH2前7个外显子的187 Kb新发重复变异(本团队2019年报道)。基因检测结果与MMR免疫组化结果一致,证实MSH2基因变异占主导。32例LS患者共检测到94个意义未明变异(VUS),其中FANCA基因VUS频率最高(25%),其次为CDH1和ATM(19%)、APC和DICER1(12%)。LLS队列所有患者均表现为微卫星高度不稳定和/或MLH1或MLH1/PMS2表达缺失,仅在非MMR基因中发现3个致病性变异:MUTYH(NM_001128425.2:c.734G>A)、FANCM(NM_020937.4:c.5101C>T)和XPA(NM_000380.3:c.820T>G)。LLS队列共检测到108个VUS,高频突变基因包括FANCA(21%)、ATM(26%)、SLX4(18%)、ALK和MSH2(15%)、WRN(12%)。散发性队列在FANCC(NM_001243743.2:c.346-1G>A)和FANCI(NM_001113378.2:c.1973delT)中发现2个致病性/可能致病性变异。在散发性队列中发现了97个VUS,高频突变基因包括SLX4、FANCA、MSH2、NF1、RECQL4(各17%)、ATM、ERCC2、NBN、XPC(各14%)等基因。仅用于研究目的时,通过VarSome Premium对所有VUS进行分类,发现3个散发性病例(S15、S3、S20)、1例LLS患者(1396)和1例LS患者(M196/01)的剪接区变异被分类为可能致病性/致病性(表2)。
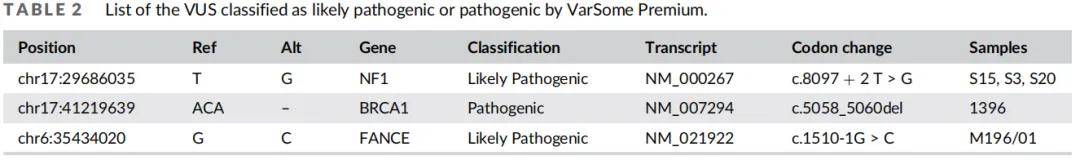
表2
LS、LLS和散发性病例与健康对照的甲基化谱比较:
为评估表观遗传对LLS表型的贡献,研究采用两步分析:样本组分析(RnBeads)和随机表观遗传突变(SEMs)分析,比较三组病例与健康对照的结果。
样本组特征:队列间差异甲基化分析
首先进行探索性主成分分析(PCA),结果显示CpG位点水平上,病例组(LS、LLS、散发性)与健康对照组无显著分离(图1A)。
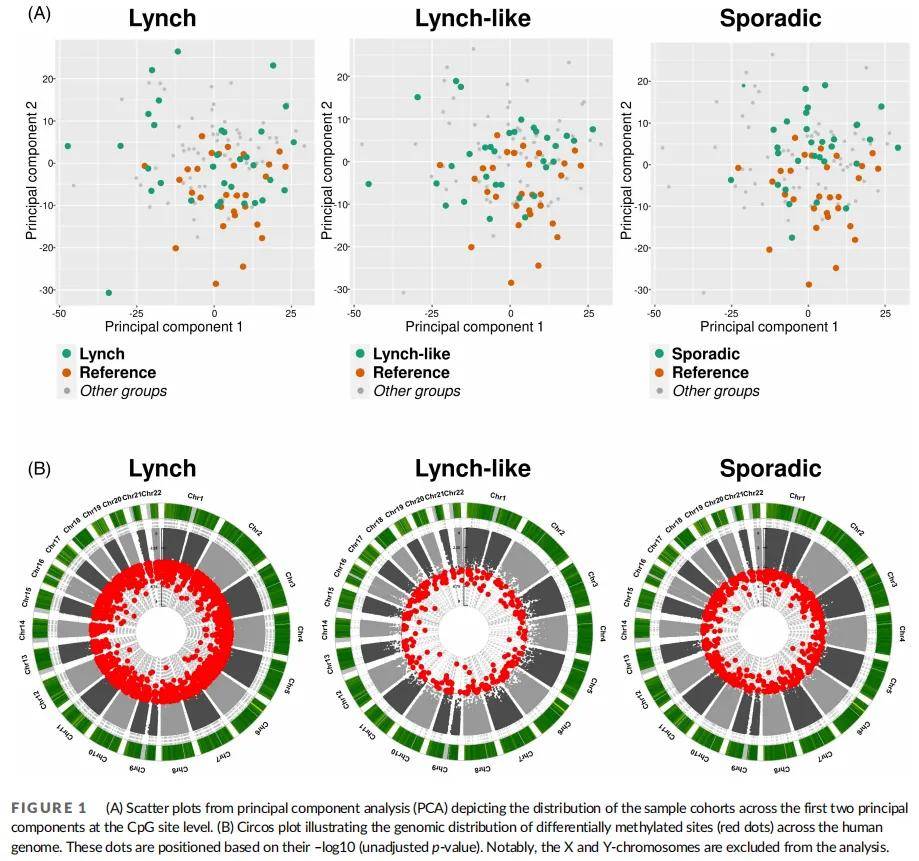
图1
通过校正潜在混杂因素(性别、年龄、细胞成分),在位点水平进行差异甲基化分析,发现LS组存在2121个差异甲基化位点(1279个高甲基化、842个低甲基化),LLS组266个(214个高甲基化、52个低甲基化),散发性组650个(64个高甲基化、587个低甲基化)(图1B)。初始分析聚焦基因位点,发现仅孤立性失调,无特定基因位点富集。为优化优先级策略,将三组基因列表与NGS panel包含的基因交叉比对:LLS组在MET和SLX4基因中存在对应变异;LS组在SLX4基因及ALK、CDC73、EZH2、FANCC、KIT、RECQL4、RUNX1、WRN基因中存在变异;散发性组在EGFR基因中存在变异。所有基因的表观遗传失调均表现为孤立位点,分散于基因体或启动子区域。区域水平(基因和启动子)未观察到显著表观遗传失调。
单病例特征:表观遗传漂移
通过检测SEMs负荷(定义为全局表观突变负荷,EML)分析表观遗传漂移,即当患者特定位点甲基化状态超出参考范围时视为发生SEMs(方法学细节见材料与方法)。健康对照组平均SEMs数量为2270(中位数:1096;IQR:906–2446),LLS组为6215(中位数:3028;IQR:1705–8160),LS组和散发性组分别为9753(中位数:4034;IQR:1879–12,803)和4699(中位数:3408;IQR:1833–6389)。经性别、年龄、细胞成分等协变量校正的多元回归模型证实,三组SEMs负荷在统计学上均较高(LS:p=3.1×10⁻⁵,LLS:p=3.8×10⁻³,散发性:p=1.9×10⁻³)(图2)。
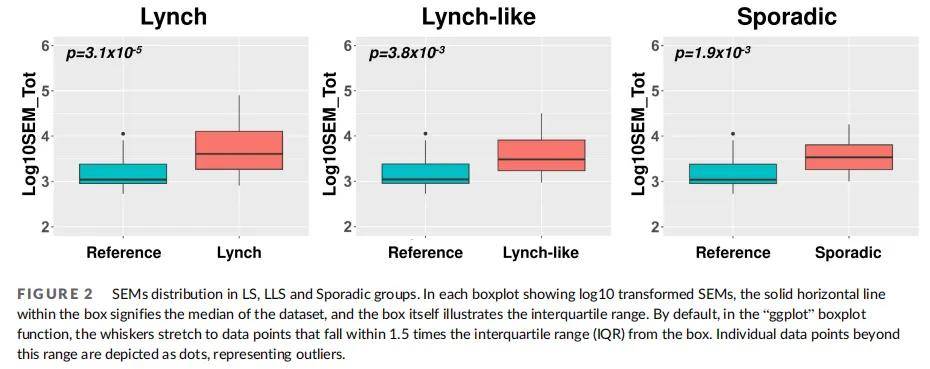
图2
然后,研究者通过确定富集基因位点的SEMs(称为表观变异),重点研究了基因水平上的SEMs负荷。为此,研究者使用了材料和方法中描述的Gentilini等开发的经过验证的方法。为了增强在三个队列中发现的解除调控基因的稳健性,研究者删除了对照队列中SEMs中发生富集的所有基因位点,产生了单一基因列表。
仅考虑高甲基化状态时,Venn分析显示LS组有1071个独特基因,LLS组593个,散发性组311个(图3A)。通过VarElect对LLS组593个独特基因进行LS表型优先级排序,发现46个与综合征直接相关的基因,其中关联度最高的基因为MLH1,其次为FAN1、EPM2AIP1、ERBB2。LS组1071个独特基因的VarElect优先级排序显示70个与综合征直接相关的基因,前四位基因为MSH2/KCNK12、FAN1、ERBB2、PTPN13,均在单例患者中呈现高甲基化,其中MSH2/KCNK12高甲基化患者同时存在MSH2基因CNV突变。散发性队列的VarElect优先级排序显示FAN1、TFCP2、PRKDC、HOXA11-AS基因分别在2、1、1、2例患者中存在高甲基化。三组患者队列的表观变异比较未显示显著差异,但发现55个共同表观变异(图3B)。VarElect对55个基因的优先级排序显示,其中5个基因与LS相关(FAN1、HOXA11-AS、OPCML、ENTPD1-AS、ERBB4),但其他基因如ASCL2和CAVIN3也在列表中。
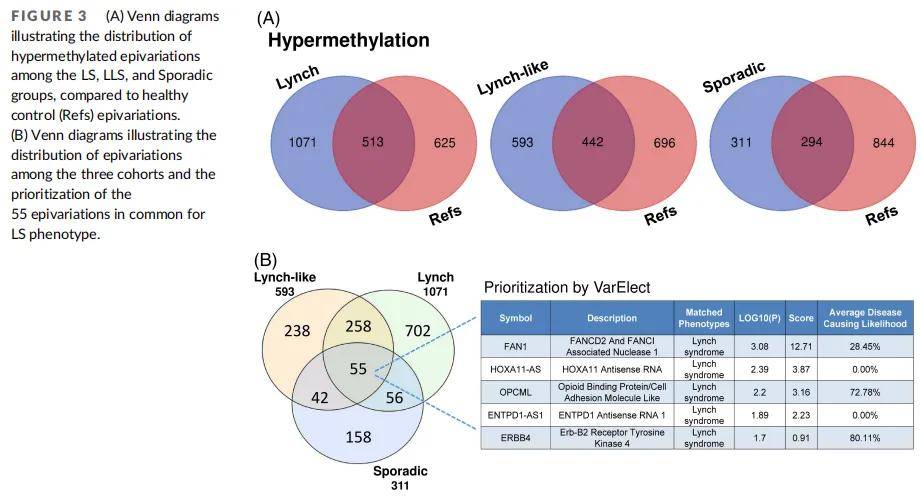
图3
讨 论
尽管经过大量研究,LLS的病因仍不明确,但其潜在原因可能包括:尚未归类为致病性的意义未明变异(VUS)、非MMR基因(如MUTYH、FANCM和XPA)的致病性突变,以及表观遗传学的作用。在此,研究者结合了94个遗传性疾病相关基因的测序,以及通过遗传咨询和基因检测确诊的LS、LLS和散发性CRC患者队列中MMR基因的CNV分析,并通过单病例甲基化分析方法评估了表观遗传学的贡献。
本研究LLS患者在发病年龄和第二、第三原发肿瘤的存在方面表现出介于LS和散发性CRC之间的临床病理特征。此外,第二原发肿瘤的部位包括结肠,但乳腺癌占比较高。
在基因水平上,研究者在非MMR基因中鉴定出三个可能被视为易感性原因的突变,并可以解释LLS患者疾病的中等外显率。其一是MUTYH中的单等位基因致病性变异。该基因的双等位基因突变易导致MUTYH相关息肉病(MAP),但在本研究病例中,患者不能归类为MAP。然而,MUTYH单等位基因突变与CRC风险增加的关联已有报道,但仍存在争议。FANCM基因已被报道与家族性乳腺癌相关。FANCM编码一种与多种DNA修复蛋白相互作用的蛋白质,对停滞复制叉的修复至关重要。XPA中的变异是一种此前从未报道过的终止密码子丢失突变,VarSome证实其具有致病性。该变异位于外显子6的聚合酶II H转录因子(TFIIH)结合位点内。XPA是一种参与核苷酸切除修复(NER)的蛋白质,通过验证DNA损伤并在修复过程中稳定DNA发挥作用。该基因变异与遗传性疾病着色性干皮病相关,但不能排除NER功能障碍(进而XPA功能障碍)可能易导致CRC的发生。此外,两名散发性患者在FANCC和FANCI中存在致病性/可能致病性变异,这些基因均参与DNA修复机制。FANCC中的变异是剪接区的功能丧失性变异,已被报道但与LS无相关性,而FANCI变异是此前从未报道过的移码缺失。通过VarSome对VUS的分析,研究者在NF1和BRCA-范可尼贫血(FA)通路(FANCE、BRCA1)中鉴定出另外三个变异(尽管有待确认),但此前未报道过与LS相关。NF1(神经纤维蛋白1)变异(c.8160+2 T>G)在ClinVar中被报告为VUS;然而,已有报道称NF1功能障碍与发生胃肠道间质瘤(GIST)、胃肠道和结直肠癌的风险增加可能相关。BRCA1是众所周知的与遗传性乳腺癌和卵巢癌(HBOC)综合征易感性相关的基因,但非移码缺失c.5058_5060del此前从未被报道过。此外,FANCE变异此前从未被报道与LS相关。然而,最近的研究强调了FA基因单等位基因突变在增加乳腺癌/卵巢癌易感性中的作用,尽管在遗传性非息肉病性结直肠癌(HNPCC)中的作用程度较小。由于研究的局限性,无法评估LLS病例中鉴定出的变异在家族成员中的存在情况,也无法进行功能研究。
为了阐明表观遗传学在LLS病例中的作用,研究者进行了甲基化表征并通过两步分析对数据进行了分析。PCA显示,在CpG位点水平和区域(基因/启动子/CpG岛),各队列与对照组之间未发现明显的表观遗传变异模式。在位点水平,分析突出了组特异性特征,使得能够鉴定出一些与NGS panel共享的基因。然而,这种失调似乎是由单个独立的变异驱动的,而不是由相邻单个失调的强烈富集模式驱动的。通过SEM分析,研究者观察到所有患者队列的表观遗传漂移显著更高。关于LLS队列,公共队列(GSE128064和GSE107353:Illumina 450K BeadChip,分别为n=112例LLS和n=41例对照)的甲基化谱证实了SEM负荷的增加(数据未显示)。这些发现支持LS、LLS和散发性患者异常积累SEM的假设。表观遗传漂移的增加可能对个体健康产生显著影响,导致基因组不稳定性增强/基因表达异常。在基因水平上,研究者在与LS相关或属于DNA修复系统的基因上鉴定出明确的表观变异。高甲基化表观变异列表的优先级排序提供了许多可能解释癌症易感性的候选基因。在LLS队列中,关联度最高的基因是MLH1,研究者在一个病例中发现其启动子区域高甲基化,该病例同时存在MUTYH致病性突变,并且表现出MLH1和PMS2表达缺失。Zyla R.及其同事曾报道过类似病例,患者最终被诊断为林奇综合征。另一个与LS关联度最高的基因是FAN1,在两个病例中高甲基化,且参与DNA修复。此外,先前的数据显示,FAN1和MLH1之间的相互作用会阻止MLH1与MSH3结合,从而抑制功能性MMR复合物的组装。列表中的第三个基因是EPM2AIP1,它与MLH1共享启动子;它与MLH1头对头定位,并以相反方向转录。事实上,EPM2AIP1和MLH1在同一患者中均发生高甲基化。前四名中的另一个基因是ERBB2,在符合Bethesda或Amsterdam II标准的被视为LS或LLS的CRC患者中突变频率较高,但表观突变的影响尚不清楚。在LS队列中,关联度最高的基因是MSH2/KCNK12,其次是FAN1、ERBB2和PTPN13。KCNK12是一个编码位于MSH2和MSH6之间的钾双孔结构域通道亚家族K成员12的基因。目前尚未报道与该综合征的直接关联,但该基因在CRC患者中常发生高甲基化。PTPN13体细胞突变已在MMR缺陷型CRC中报道,但其作用尚不清楚。在散发性队列中,关联度最高的基因是FAN1、TFCP2、PRKDC、HOXA11-AS。PRKDC产生一种参与细胞周期控制的蛋白质,能够直接与MSH2相互作用。它还参与DNA非同源末端连接(NHEJ),这是双链断裂(DSB)修复和V(D)J重组所必需的。最近,PRKDC基因的创始人突变与CRC中突变负荷增加相关,因此该基因已被指出是肿瘤异质性的新驱动因素。HOXA11-AS的过表达与CRC进展和预后不良相关,并可能促进转移,但尚未有关于其高甲基化的数据报道。
基于表观变异与疾病的关联及其在各队列中的共性进行分析,研究者发现所有三个队列中均有患者存在FAN1、CAVIN3和ASCL2的表观变异。CAVIN3是一种假定的抑癌基因,已发现在许多癌症中失活。其在正确DNA修复中起关键作用,因为其蛋白质与BRCA1相互作用,稳定BRCA1并确保其正常运作。当CAVIN3不表达时,细胞中BRCA1的水平降低,如修复DNA的能力下降。ASCL2是最近一项关于LS表观遗传特征的研究中报道的基因。作者描述了包括ASCL2在内的干细胞标记基因启动子中高水平的H3K27me3,表明其高甲基化。需要进一步的研究来了解表观变异在蛋白质表达、这些修饰的遗传性及其外显率方面的贡献程度。
本研究在MMR基因和FA通路中鉴定出致病性变异,这些变异可以将部分(而非全部)LLS患者重新归类为LS病例。散发性患者的遗传分析有助于鉴定可能增加癌症易感性的致病性变异。本研究结果强调了在诊断过程中使用广泛的基因panel以避免误分类的重要性,从而防止患者无法加入家族预防和监测系统。此外,研究表明遗传分析也应扩展到散发性癌症患者,以避免漏诊。尽管缺乏功能验证和家族外显率数据,本研究结果提供了新的见解,值得在更大的队列中进行验证,以帮助验证有害变异和非MMR特异性基因高甲基化在疾病发生中的作用和风险,以及它们纳入诊断性检测的潜力。
参考文献:
Pirini F, Calzari L, Tedaldi G, et al. Comprehensive genetic and epigenetic characterization of Lynch-like syndrome patients. Int J Cancer. Published online April 21, 2025. doi:10.1002/ijc.35451